
Fibrosi cistica, nuove speranze di cura

Il nostro menù del giorno 03_11_2021
3 Novembre 2021
APGAR – primo valore della nascita in cartella clinica
3 Novembre 2021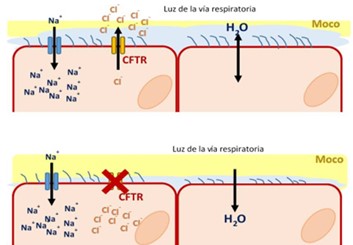
Gli antichi romani ne erano già a conoscenza, era chiamata: la malattia del bacio salato. Fino agli anni ‘40 la diagnosi della malattia equivaleva a una condanna, la speranza di vita era di pochi anni a volta di mesi. Stiamo parlando della fibrosi cistica o mucoviscidosi, una malattia genetica che colpisce le ghiandole produttrici di sostanze come il muco e il sudore. Fortunatamente la ricerca ha permesso la realizzazione di nuovi farmaci permettendone una qualità della vita non solo migliore ma anche di raggiungere un’età molto più avanzata rispetto a un tempo.
Fibrosi cistica: la malattia
La fibrosi cistica è causata a mutazioni nel gene CFTR, responsabile della codifica della proteina attiva sul passaggio di acqua e sali all’interno e all’esterno delle cellule. Il risultato è la produzione di muco molto denso e sudore ricco di sali, con livelli di serietà diversi a seconda delle mutazioni, in totale sono circa mille. Quindi le persone con fibrosi cistica producono un muco denso e appiccicoso al punto che possono ostruire le vie respiratorie e i dotti dell’apparato digerente che permettono agli enzimi del pancreas di raggiungere l’intestino tenue per contribuire alla digestione. Le conseguenze più importanti sono: problemi digestivi e di assorbimento, aumento del rischio di infezioni batteriche, danni ai polmoni, problemi intestinali e addominali. La malattia espone anche a rischio di osteoporosi e di problemi di infertilità.
I sintomi della malattia fibrocistica
I sintomi della fibrosi cistica cambiano da caso a caso e spesso mutano con il passare degli anni. Spesso nei bambini piccoli compare stitichezza ostinata già alla nascita e sudorazione salata. Con il tempo compaiono poi disturbi delle vie respiratorie con infezioni ricorrenti e resistenti agli antibiotici. Possono comparire malattie del fegato e della cistifellea, a livello dell’apparato digerente: diarrea o feci dall’aspetto oleoso e nauseabonde, blocchi intestinali, flatulenza eccessiva e stitichezza grave associati a dolori addominali, carenze nutrizionali che ostacolano la crescita e l’aumento di peso, pancreatite, prolassi rettali, malattie epatiche, diabete e calcoli alla cistifellea. La prevenzione della malattia è possibile solo effettuando analisi genetiche su entrambi i partner. La malattia compare solo se entrambe le copie di CFTR presenti in una persona sono mutate. Se tutti e due i genitori sono portatori sani della malattia con una sola copia di CFTR mutata, per ogni figlio la probabilità di essere affetto dalla malattia è del 25%, quella di essere portatore è del 50% e quella di avere entrambe le coppie sane di CFTR del 25%. Si può poi effettuare l’analisi genetica sul materiale prelevato in gravidanza attraverso amniocentesi o villocentesi, per scoprire se il feto è portatore di malattia.
Le nuove possibilità di cura
Secondo i dati del Registro, i malati in Italia sono 6.000, di cui circa la metà ha meno di 18 anni. Fino a pochi anni fa, erano disponibili solo farmaci sintomatici, permettendo di migliorare le aspettative di vita: gli antibiotici, i fluidificanti, gli antinfiammatori e gli enzimi pancreatici. È del 1989, l’identificazione del gene responsabile dell’anomalia della proteina CFTR che regola gli scambi degli ioni di sodio e cloro a livello degli epiteli ghiandolari e che determina la malattia. Questa nuova scoperta ha portato alla formulazione dei modulatori della proteina CFTR cambiando l’aspettativa di vita. Da 2015 esistono farmaci mirati per la proteina CFTR, quindi oggi oltre il 50% dei bambini nati da quell’anno in poi, anno in cui sono stati usati per la prima volta i nuovi farmaci modulatori della proteina CFTR, riesce a sopravvivere oltre i 50 anni. Un risultato inaspettato se si pensa che quando è stato scoperto il fenotipo clinico, 83 anni fa, i bambini morivano poco dopo la diagnosi.
Sahalima Giovannini
Sostieni Guidagenitori.it
Il Covid-19 ha colpito al cuore anche l’economia, tutto si è fermato, pubblicità compresa, l’unica forma di sostentamento per fare e diffondere l’informazione medico-scientifica, obiettivo principale di Guidagenitori.it
I nostri giornalisti, tecnici informatici e tutti gli altri operatori che sorreggono il giornale, continuano a svolgere regolarmente il lavoro per offrire gratuitamente i servizi editoriali, nonostante le difficoltà economiche. Ecco perché il vostro contributo è prezioso.
